
متلازمة دي جورج، المعروفة أيضاً باسم متلازمة حذف 22q11.2، هي اضطراب جيني نادر يحدث نتيجة فقدان جزء صغير من كروموسوم 22. تعد هذه المتلازمة مصدر قلق وتحدياً للأفراد الذين يعانون منها ولعائلاتهم، حيث تؤثر على عدة جوانب من حياتهم اليومية. سنقوم في هذا المقال بالتعرف على تفاصيل هذا الاضطراب، وتأثيره على الأفراد المصابين به وعلى محيطهم، بالإضافة إلى أحدث التطورات في مجال علاجه وإدارته.

يغطي مصطلح متلازمة حذف 22q11.2 المصطلحات التي كان يُعتقد أنها حالات مختلفة، ويشمل ذلك متلازمة دي جورج والمتلازمة الشراعية القلبية الوجهية velocardiofacial syndrome والاضطرابات الأخرى التي تشارك نفس السبب الجيني بالرغم من وجود اختلاف بسيط في السمات.
تتضمن المشاكل الصحية الأكثر شيوعًا المرتبطة بمتلازمة حذف 22q11.2 عيوب القلب وضعف وظيفة الجهاز المناعي والحنك المشقوق والمضاعفات مرتبطة بانخفاض مستويات الكالسيوم في الدم وتأخر في النمو مع مشكلات سلوكية وعاطفية…
تختلف أرقام الأعراض المرتبطة بمتلازمة حذف 22q11.2 وشدتها. ومع ذلك، يحتاج كل شخص تقريبًا مصاب بهذه المتلازمة إلى العلاج من متخصصين في مختلف المجالات.
سيكون هذا المقال للحديث عن كل ما يخص هذه المتلازمة، ابتداءًا من أسبابها وأعراضها وصولًا إلى تشخيصها وعلاجها.
محتويات المقال :
انتشار متلازمة دي جورج:
تصيب المتلازمة ما يقدر بـ 1 من كل 4000 إلى 6000 ولادة حية. وهي نسبة عالية مقارنة بباقي الأمراض. حيث تعتبر النسبة الأعلى بين باقي الأمراض الوراثية التي تحدث بسبب حذف microdeletion syndrome.
[1]
أسبابها:
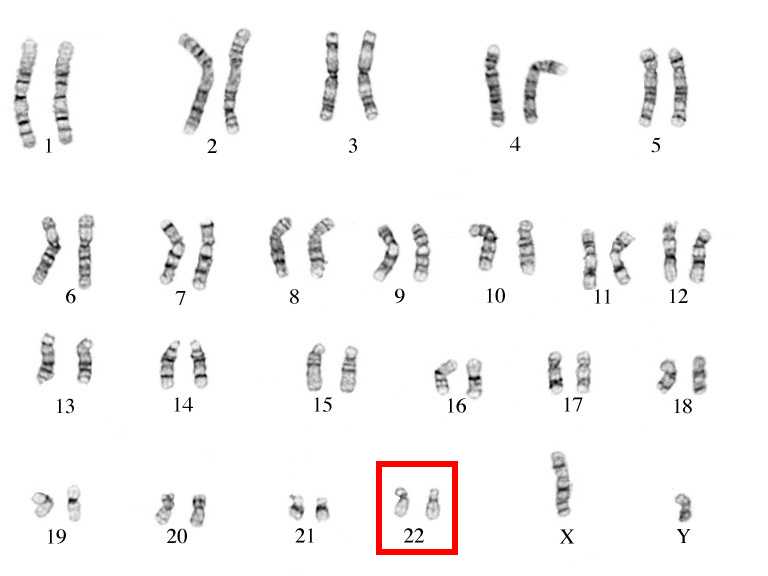
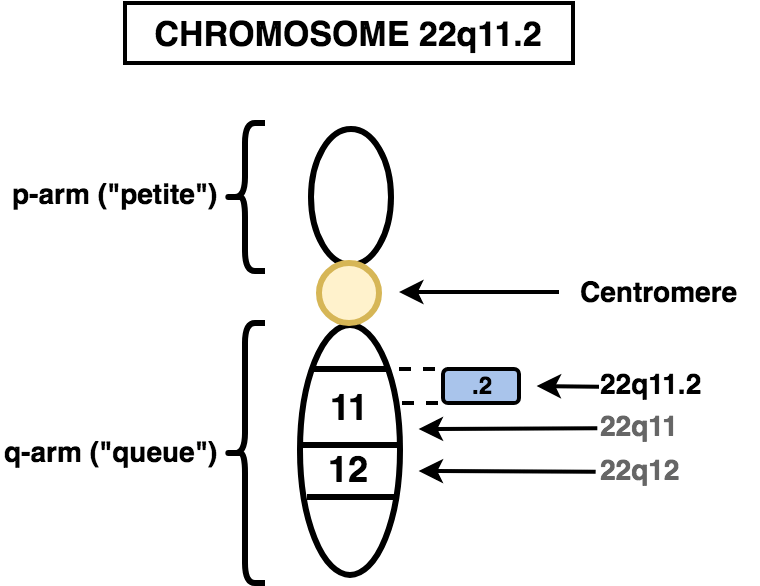
بدايةً نعلم أن كل شخص لديه نسختين من الكروموسوم 22، حيث تورّث واحدة من الأب والأخرى من الأم. وإذا كان الشخص مصابًا بمتلازمة دي جورج، فإن نسخة واحدة من الكروموسوم 22 تفتقر إلى جزء يحتوي على ما يقدر بـ 30 إلى 40 جين (حوالي 3 ملايين زوج قاعدة base pairs). ولم يتم تحديد العديد من هذه الجينات بوضوح ولم تكن مفهومة جيدًا. تعرف منطقة الكروموسوم 22 التي تم حذفها باسم 22q11.2.
عادةً ما يحدث حذف الجينات من الكروموسوم 22 كحدث عشوائي في الحيوان المنوي للأب أو في بويضة الأم، أو قد يحدث مبكرًا خلال نمو الجنين. ونادراً ما يكون الحذف حالة وراثية يتم نقلها إلى طفل من أحد الوالدين الذي لديه أيضًا عمليات حذف في الكروموسوم 22 ولكن قد لا تظهر عليه أعراض.
وبالنسبة لوراثة المتلازمة، فهي من نوع الـ Autosomal Dominant: أي أن إصابة أحد الوالدين بها، يعطي احتمالية 50% لإصابة كل جنين بالمتلازمة. [1][2]

الأعراض:
قد تتفاوت علامات متلازمة دي جورج وأعراضها في نوعها وحدتها بناءً على الأجهزة المصابة في الجسم ومدى شدة الإصابات. ,وربما تكون بعض العلامات والأعراض ظاهرة عند الولادة، ويمكن أن لا يظهر بعضها حتى أواخر مرحلة الرضاعة أو أوائل مرحلة الطفولة. [2][3]
أهم العلامات والأعراض:
- النفخة القلبية وزرقة البشرة نتيجة لقلة انتشار الدم الغني بالأكسجين (الزراق) بسبب الإصابة بعيب في القلب.
- العدوى المتكررة.
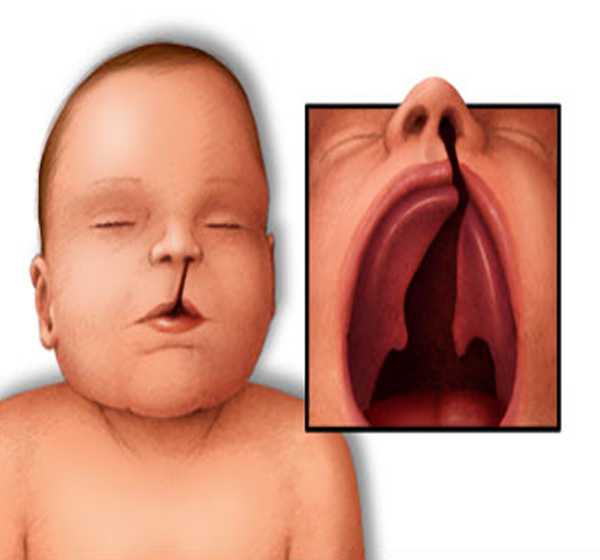
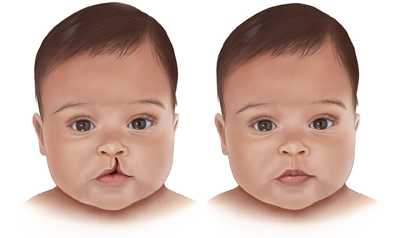
- سمات وجه معينة، مثل ذقن غير مكتملة النمو، أو أذنان منخفضتان، أو عينان متباعدتان أو ثلم ضيق بالشفة العليا – الشفة الأرنبية-
- فجوة في سقف الفم (الحنك المشقوق palate defect) أو مشكلات أخرى في الحنك.
- صعوبة التغذية أو عدم زيادة الوزن أو مشاكل في الجهاز الهضمي.
- صعوبات في التنفس.
- توتر عضلات ضعيف.
- تأخر النمو، مثل تأخر التدحرج أو الجلوس أو غيرها من المراحل الأساسية في نمو الرضيع.
- تأخر تطور الكلام أو الكلام المصحوب بغنة أنفية.
- إعاقات التعلم أو تأخره.
- مشكلات سلوكية.
متى تزور الطبيب
توجد حالات أخرى يمكن أن تسبب علامات وأعراض مشابهة لمتلازمة حذف 22q11.2. وبالتالي يجب الحصول على تشخيص دقيق وعاجل إذا ظهر على طفلك أي من العلامات أو الأعراض سابقة الذكر.
قد يشك الطبيب في الإصابة بمتلازمة حذف 22q11.2 في الأوقات التالية:
- عند الولادة. إذا بدت حالات معينة واضحة عند الولادة، مثل عيب خطير في القلب أو الحنك المشقوق أو عدة عوامل أخرى تشبه ملازمة حذف 22q11.2، فمن المرجح أن تبدأ الاختبارات التشخيصية قبل أن يغادر الطفل المستشفى.
- في زيارات العناية بالطفل. قد يشتبه طبيب العائلة أو طبيب الأطفال في وجود هذا الاضطراب بسبب مجموعة من الأمراض أو الاضطرابات التي تتضح مع مرور الوقت. وقد ينتبه الطبيب لوجود مشكلات أخرى أثناء زيارات فحص صحة المولود المقررة بانتظام أو خلال الفحوصات السنوية للطفل. [2][1]
المضاعفات:
تلعب أجزاء الصبغيات 22 المحذوفة في متلازمة دي جورج دورًا في تطور عدد من أجهزة الجسم. وكنتيجة لذلك، يُمكن أن يؤدي الاضطراب إلى حدوث طفرات عديدة في أثناء نمو الجنين. وتتضمن المشكلات الشائعة ما يلي:
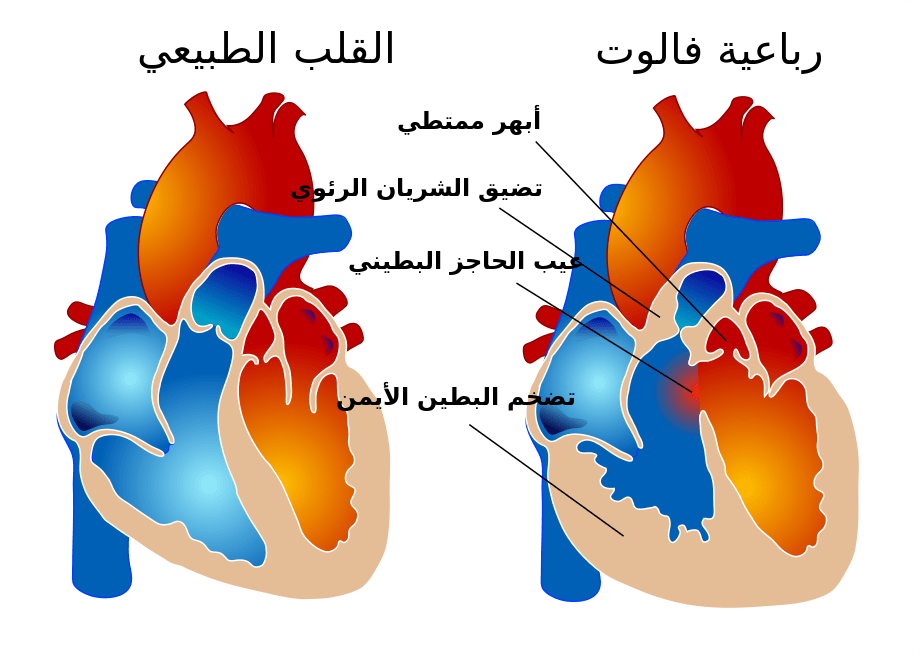
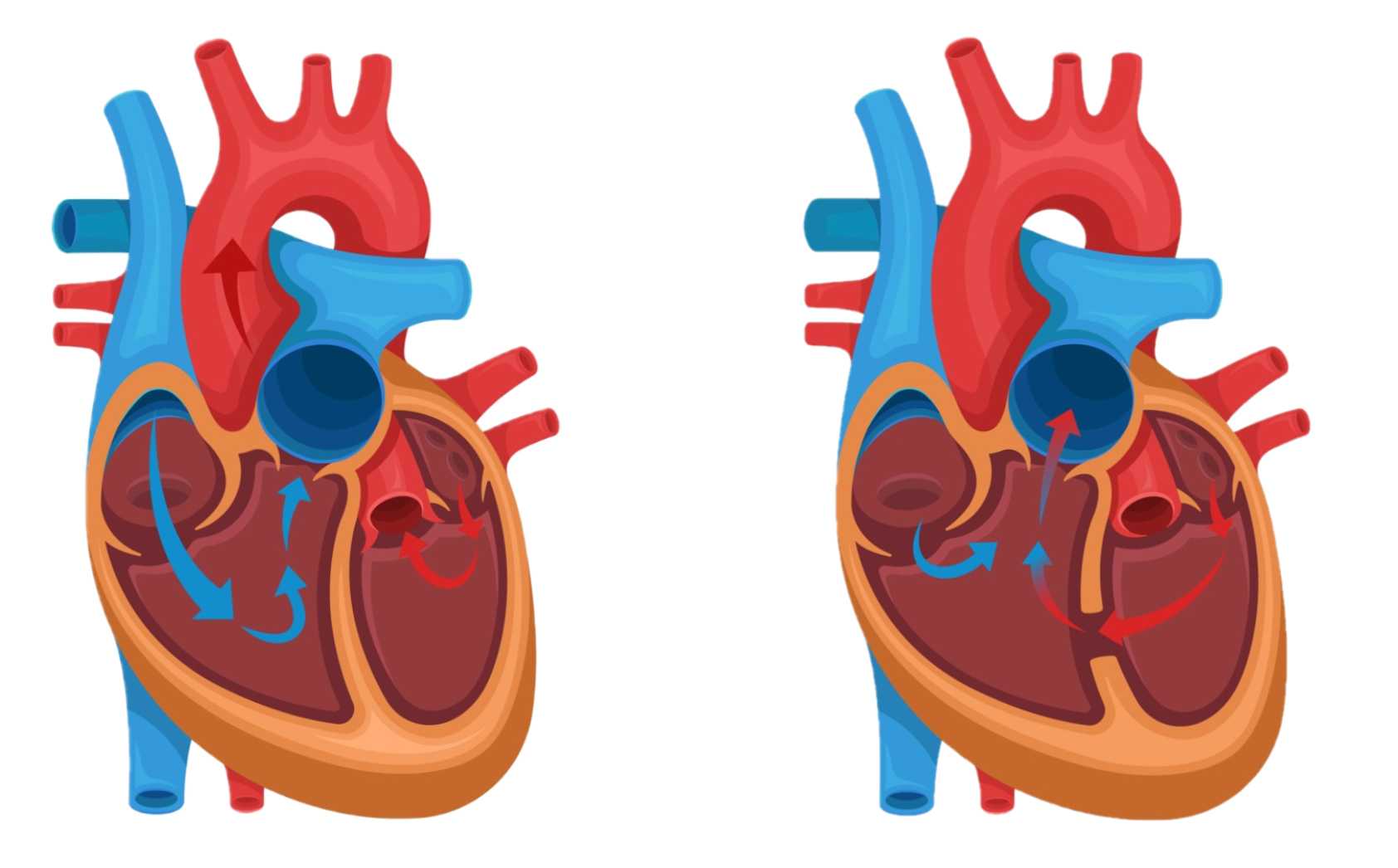
- عيوب القلب: وغالبًا ما تسبب متلازمة حذف 22q11.2 عيوبًا في القلب قد ينجم عنها عدم كفاية إمدادات الدم الغني بالأكسجين. على سبيل المثال، قد تتضمن التشوهات وجود ثقب بين الحجرات السفلية في القلب (خلل في الحاجز البطيني ventricular septal defect) أو وجود وعاء كبير فقط؛ بدلاً من وعائين، يؤدي إلى خارج القلب (الجذع الشرياني truncus arteriosus) أو مجموعة من أربعة هياكل قلبية غير طبيعية (رباعية فالو tetralogy of fallot).
- قُصورُ الدُّرَيْقات: تقوم الغدد فوق الدرقية الأربعة بتنظيم مستويات الكالسيوم والفوسفور في الجسم. يُمكن أن تؤدي متلازمة حذف 22q11.2 إلى تصغير الغدد فوق الدرقية أكثر من الطبيعي وهي الغدد التي تفرز مقدارًا صغيرًا جدًا من الهرمون الجار درقي (PTH)، ما يؤدي إلى الإصابة بقُصور الدُّرَيْقات congenital hypoparathyroidism. وينجم عن هذه الحالة وجود مستويات منخفضة من الكالسيوم ومستويات مرتفعة من الفوسفور في الدم.
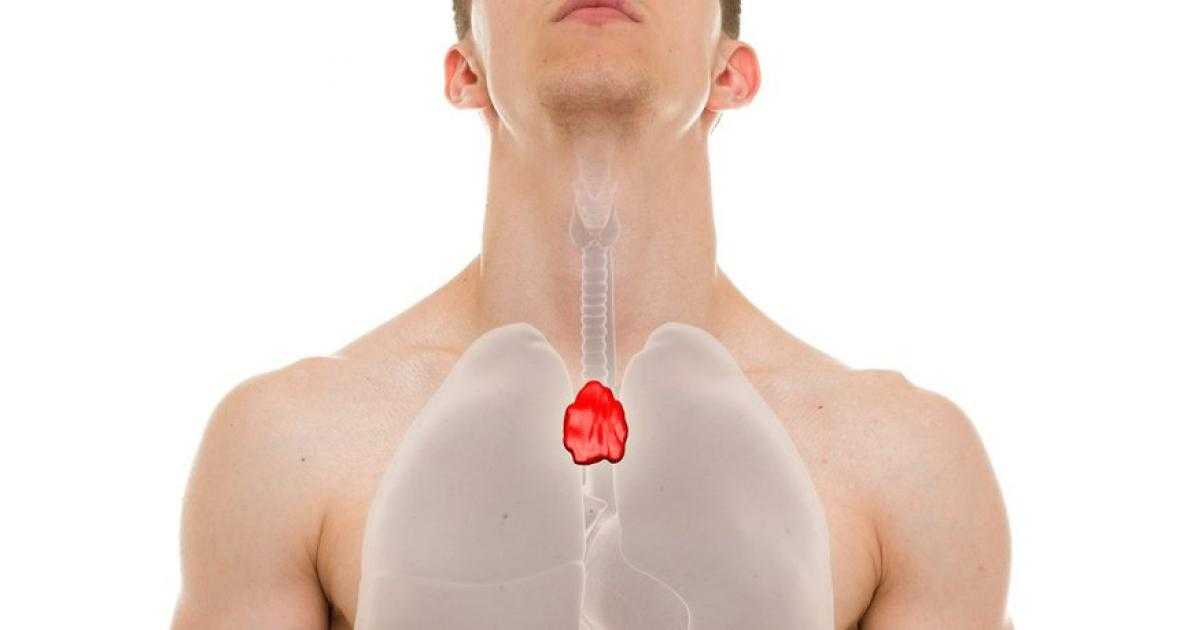
- خلل في الغدة الزعترية: تنضج الخلايا التائية؛ نوع من خلايا الدم البيضاء، في الغدة الزعترية الموجودة أسفل عظمة الصدر. وتعتبر الخلايا التائية الناضجة ضرورية للمساعدة في مقاومة العدوى. بالنسبة إلى الأطفال المصابين بمتلازمة حذف 22q11.2، قد تكون الغدة الزعترية صغيرة أو مفقودة thymic aplasia، الأمر الذي يؤدي إلى ضعف وظيفة الجهاز المناعي وحدوث عدوى شديدة بشكل متكرر.
- حنك مشقوق: إن الحنك المشقوق؛ فتحة (شق) في سطح الفم (الحنك) cleft palate، سواء وجدت شفة مشقوقة أو لا، يُعتبر حالة شائعة من متلازمة حذف 22q11.2. وقد تصبح عملية البلع أو إصدار أصوات معينة في أثناء الكلام صعبة بسبب وجود تغييرات غير طبيعية أخرى غير واضحة بالحنك.
- ملامح وجه متميزة: ربما يظهر عدد من السمات المحددة بوجه بعض الأشخاص المصابين بمتلازمة حذف 22q11.2. وهذه السمات تتضمن وجود أذنين صغيرتين متجهتين للأسفل أو جفون الأعين صغيرة الاتساع (شقوق جفنية) أو الأعين المقنعة أو وجه طويل نسبيًا أو طرف أنف ضخم (بصلية الشكل) أو ثلم صغير أو مسطح بالشفة العليا.
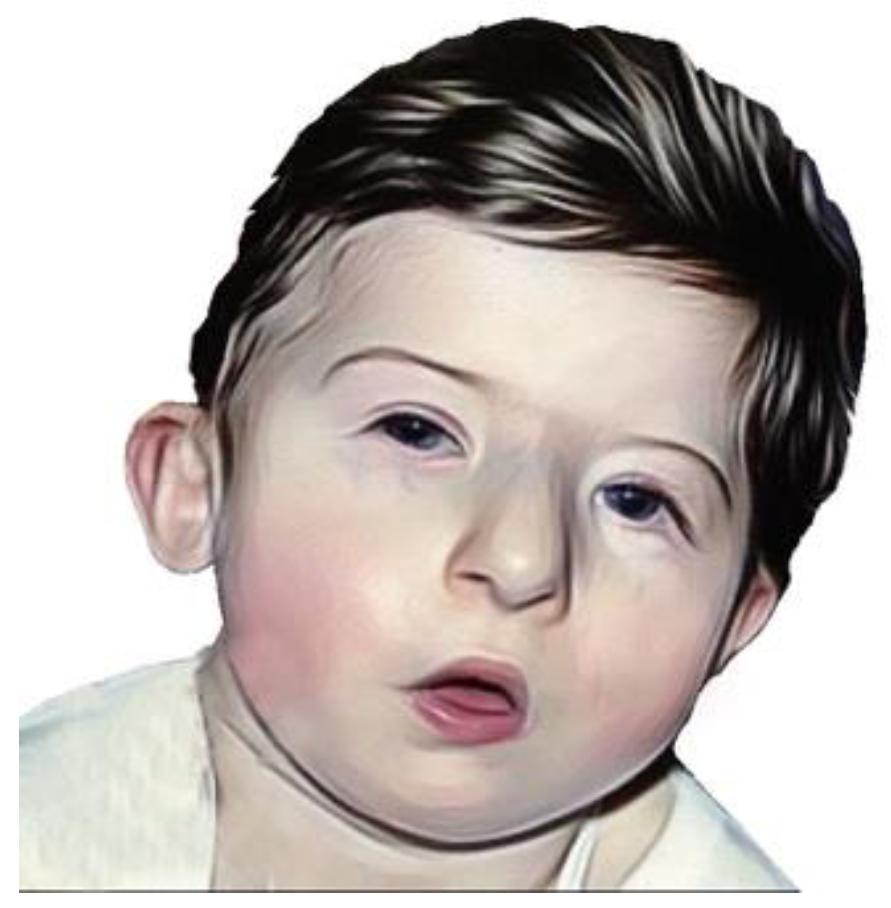
- مشكلات في التعلم والسلوكيات والصحة العقلية: قد تسبب متلازمة حذف 22q11.2 مشكلات في نمو الدماغ وعمله، الأمر الذي يؤدي إلى حدوث مشاكل في التعلم والعلاقات الاجتماعية والنمو والسلوكيات. إن تأخر تطور الكلام وصعوبات التعلم لدى الأطفال من الحالات الشائعة. فبعض الأطفال يُصابون باضطراب نقص الانتباه مع فرط النشاط (ADHD) أو اضطراب طيف التوحد. وفي مرحلة لاحقة في الحياة، تزيد خطورة الإصابة بالاكتئاب واضطرابات القلق واضطرابات صحية أخرى.
- اضطرابات المناعة الذاتية: كما يُمكن أن يكون الأشخاص الذين كانوا يعانون من ضعف وظيفة الجهاز المناعي في طفولتهم؛ بسبب صغر أو فقدان الغدة الزعترية، أكثر عرضة للإصابة باضطرابات المناعة الذاتية، مثل التهاب المفاصل الروماتويدي أو داء غريفز (الدُّرَاق الجُحُوظِيّ).
- مشاكل أخرى. قد يقترن عدد كبير من الحالات الطبية بمتلازمة حذف 22q11.2، مثل ضعف السمع وضعف البصر ومشاكل في التنفس وضعف في وظيفة الكلى وقصر القامة نسبيًا بالنسبة إلى طول أفراد العائلة. [2]
الوقاية
في بعض الحالات، تُمرر متلازمة دي جورج من الوالد المصاب إلى الطفل. إذا كنت قلقًا بشأن تاريخ عائلي لمتلازمة حذف 22q11.2، أو إذا كان لديك بالفعل طفل مصاب بالمتلازمة، فقد ترغب في استشارة طبيب متخصص في الاضطرابات الوراثية (أخصائي طب الوراثيات) أو استشاري الوراثيات للمساعدة في التخطيط للحمل في المستقبل.
التشخيص:
يستند تشخيص متلازمة دي جورج في المقام الأول على الاختبارات المعملية الذي يمكنه الكشف عن الحذف في الصبغي 22. قد یطلب طبیبك ھذا الاختبار إذا کان لدى طفلك:
- مجموعة من المشكلات أو الحالات الطبية التي تشير إلى الإصابة بالمتلازمة
- عيب قلبي، لأن بعض عيوب القلب ترتبط عادة مع متلازمة حذف 22q11.2
ومن أهم التقنيات المستخدمة في التشخيص هي:
الوميض الصبغي بتهجين الفلوريسنت fluorescent in situ hybridization -FISH والذي يبين الحذف الجزئي لمقطع بالصبغي 22 حيث يتم تشخيص الحذف الجزئي للذراع الطويلة في هذا الكروموسوم. [2][1]
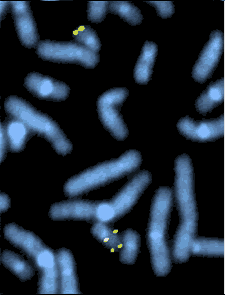
كما يمكن عمل الفحص الوراثي بعد عمل انتساخ للحامض النووي عن طريق التفاعل المتسلسل المتضاعف (polymerase chain reaction (pcr.
ملاحظة: عندما تكون الأعراض والعلامات موجودة دون وجود الحذف الجزئي بالصبغي 22, تكون أسباب أخرى هي التي تفسر وجود الأعراض, مثل وجود نقص بالصبغي 10, أو التعرض قبل الولادة للكحول, أو للأيزوتريتينون, أو ارتفاع مستوى السكر بالدم.
يمكن أيصًا استخدم قياس الانسياب الخلوي flow cytometry لتقدير عدد الخلايا التائية, والاستجابات التكاثرية لمحدثات الاستحالة أو التفتل mitogens والمستضدات antigens.
إضافة إلى إمكانية استخدام فحوض الأشعة كالرنين المغناطيسي في تأكيد وتقييم الحالة…
العلاج:
بالرغم من عدم وجود علاج لمتلازمة دي جورج، إلا أن العلاجات يمكنها عادةً علاج المشكلات الخطيرة، مثل عيب القلب أو الحنك المشقوق. جيث يمكن معالجة المسائل الصحية والمشاكل النمائية، أو مشاكل الصحة العقلية أو المشاكل السلوكية الأخرى أو متابعتها وفقًا للحاجة.
ربما تتضمن العلاجات (العلاج المخصص) تدخلات لعلاج الأمور التالية:
- قُصورُ الدُّرَيْقات. يمكن التغلب على قُصورُ الدُّرَيْقات عادةً من خلال مكملات الكالسيوم ومكملات فيتامين (د).
- عيوب القلب. تتطلب معظم عيوب القلب جراحة بعد الولادة مباشرة لعلاج القلب وتحسين مستوى وصول الدم الغني بالأكسجين.
- قصور وظيفة الغدة الزعترية. إذا كان طفلك يعاني من بعض القصور في وظيفة الغدة الزعترية، فربما تكون حالات العدوى متكررة، ولكن دون وجود خطورة بالضرورة. يتم علاج حالات العدوى المذكورة — نزلات البرد والتهابات الأذن عادةً — بوجه عام كما هو الحال مع أي طفل. يتبع معظم الأطفال المصابين بقصور وظيفة الغدة الزعترية جدول التطعيمات الطبيعي. بالنسبة لمعظم الأطفال الذين يعانون من قصور وظيفة الغدة الزعترية بصورة معتدلة، يتحسن أداء الجهاز المناعي مع التقدم في العمر.
- خلل الغدة الزعترية الشديد. إذا كان خلل الغدة الزعترية شديدًا أو في حالة عدم وجود غدة زعترية، فيكون طفلك معرضًا لمخاطر الإصابة بمجموعة من الأمراض الخطيرة. يتطلب العلاج زراعة نسيج الغدة الزعترية، أو خلايا متخصصة من النخاع العظمي أو خلايا الدم المتخصصة المقاومة للأمراض.
- حنك مشقوق. يمكن علاج الحنك المشقوق أو غير ذلك من اضطرابات الحنك والشفة عادةً من خلال التدخل الجراحي.
- التطوير الشامل. من المحتمل أن يستفيد طفلك من مجموعة من العلاجات، بما في ذلك العلاج التخاطبي، والعلاج المهني والعلاج النمائي. في الولايات المتحدة، تتوفر برامج التدخل المبكر التي توفر الأنواع المذكورة من العلاج من خلال الإدارة الصحية بالمدينة أو الولاية.
- رعاية الصحة النفسية. ربما يوصى بالعلاج إذا تم تشخيص حالة طفلك فيما بعد على أنها إصابة باضطراب نقص الانتباه مع فرط النشاط (ADHD)، أو اضطراب طيف التوحد، أو الاكتئاب، أو غير ذلك من اضطرابات الصحة العقلية أو الاضطرابات السلوكية.

- إدارة الحالات الأخرى. ربما تتضمن الحالات المذكورة علاج المسائل المتعلقة بالتغذية والنمو، والمشاكل السمعية أو البصرية، وغير ذلك من الحالات الطبية. [3][2][1]
ملاحظة:
توجد محاولات ناجحة لزراعة الغدة التيموسية للأطفال الذين تكون عندهم الغدة غير موجودة, حيث أنه أثناء عمل جراحة للقلب, يؤخذ جزء من الغدة التيموسية من طفل حديث الولادة لأسرة متبرعة, وبعد فحصها والتأكد من خلوها من الأمراض, يتم زرع أجزاء صغيرة منها بعضلات الساق, وفي دراسة تم إجراؤها في شهر أغسطس سنة 2003 تبين أن جميع الأطفال الذين تم علاجهم بهذه الطريقة, قد حدث عندهم تطور لجهاز مناعي فعال, وهذه الأبحاث تعطى أمل, أن الأطفال المتأثرين بشدة بتشوه دي جورج من الممكن أن يعيشون حياة صحية.
في أغلب الحالات يجب تجنب التطعيم للأطفال المصابين بتشوه دي جورج بلقاحات الفيروسات الحية.
مصير المرضى:
يعتمد مصير المرض على مدى الإصابة ومدى اعتلال القلب والجهاز المناعي.
أغلب الأشخاص الذين يعيشون يكون عندهم صعوبات بسيطة في التعلم ويكون نموهم طبيعي.
الأطفال المتأثرين بشدة بالمتلازمة يكون عندهم مشاكل شديدة, مثل أمراض القلب أو غياب الغدة التيموسية, ورغم العلاج فإنهم لا يعيشون أكثر من سنوات قليلة.
أمراض وراثية أخرى ذات صلة:
مرض ترسب الأصبغة الدموي الوراثي
مراجع ومصادر:
1- ncbi
2- omim
3- medline plus
Author: Mohammad Shawerdi
طالب علم، مهتم بالمعرفة والثقافة، مع التخصص بالمجال الصحي-الطبي المتعلق بالتحاليل الطبية.
سعدنا بزيارتك، جميع مقالات الموقع هي ملك موقع الأكاديمية بوست ولا يحق لأي شخص أو جهة استخدامها دون الإشارة إليها كمصدر. تعمل إدارة الموقع على إدارة عملية كتابة المحتوى العلمي دون تدخل مباشر في أسلوب الكاتب، مما يحمل الكاتب المسؤولية عن مدى دقة وسلامة ما يكتب.



التعليقات :